
Zaburzenia ze spektrum autyzmu (ASDs, ang. autism spectrum disorders) są jedną z najczęściej występujących grup zaburzeń neurorozwojowych i dotyczą około 0,6-1% ogólnej populacji dzieci. Autyzm klasyczny będący częścią zaburzeń ASD dotyczy ok. 0,2-0,3% dzieci. Zaburzenia charakteryzują się upośledzeniem komunikacji i interakcji społecznych oraz ograniczonym, powtarzającym się i stereotypowym zachowaniem. Szacuje się, że zaburzenia ASD dotyczą łącznie około 21,7 milionów ludzi na świecie, w tym obejmują jedno na siedemdziesiąt dzieci w wieku szkolnym. Mechanizm powstawania ASD jest wciąż mało poznany, ale wiadomo, że w etiologii tych zaburzeń istotną rolę odgrywają zarówno czynniki genetyczne jak i środowiskowe.
Choroby z tzw. spektrum autystycznego (autyzm, zespół Retta, zespół Aspergera, inne zaburzenia rozwojowe i hiperkinetyczne), których wspólną cechą jest zaburzenie w komunikacji werbalnej i pozawerbalnej dziecka oraz trudności w rozumieniu zjawisk społecznych, są coraz częściej obserwowane i rozpoznawane – także w Polsce. Wśród środowiska lekarskiego trwa dyskusja o przyczynach – każdy rok przynosi nowe doniesienia.
Terapia objawowa prowadzona metodami interwencji psychoterapeutycznej oraz farmakologiczna opierają się na założeniu, że przyczyną autyzmu jest zaburzenie na tle neurorozwojowym. Metody te mają jednak ograniczoną skuteczność.
Coraz większym uznaniem cieszą się badania dowodzące, że powodem autyzmu są zaburzenia metaboliczne w obszarze metabolizmu komórkowego skutkujące obniżoną zdolnością komórek – szczególnie nerwowych – do produkcji wysokoenergetycznych cząsteczek będących źródłem energii potrzebnej dla funkcjonowania organizmu.
Centralnymi pojęciami są stres azotowy i stres oksydacyjny.
Autyzm jest jednostką chorobową doskonale ilustrującą pojęcie ekwifinalności – wiele różnych przyczyn może prowadzić do tego zespołu. To czy dziecko zostanie nim dotknięte zależy od jego podatności (polimorfizmy genowe) oraz od zetknięcia z potencjalnie niebezpiecznymi czynnikami: występuje ogromna zmienność osobnicza w zdolności tolerowania różnych czynników środowiskowych – niektóre dzieci reagują patologicznie na czynniki, które większości nie stwarzają problemu.
Do czynników mogących mieć wpływ na powstanie choroby zalicza się infekcje bakteryjne i wirusowe, dysbiozę w obrębie przewodu pokarmowego, nadwrażliwości pokarmowe, względne niedobory mikroelementów i witamin, zatrucia metalami ciężkimi.
Wszystko powyższe wymaga dokładnej i wszechstronnej diagnostyki. Tylko w ten sposób możemy wykryć potencjalny deficyt metaboliczny a niekiedy i przyczynę, i wdrożyć skuteczne leczenie. Tylko usunięcie przyczyn somatycznych i następnie terapia psychoterapeutyczna mogą doprowadzić do trwałej poprawy. Jeśli przyczyną zaburzeń jest patologiczna reakcja na bodźce środowiskowe diagnostyka pomoże w określeniu czego należy unikać w przyszłości aby nie dopuszczać do zaostrzeń choroby.
Od teraz, wszystkie badania dotyczące diagnostyki autyzmu dostępne są w ofercie ALAB laboratoria. Nasza oferta to specjalistyczne pakiety badań, stosowane przez lekarzy zajmujących się autyzmem od strony metabolicznej, pomagające w określeniu przyczyn choroby.
| PONIŻEJ PREZENTUJEMY NAJCZĘŚCIEJ WYKONYWANE BADANIA DOSTĘPNE W RAMACH DIAGNOSTYKI AUTYZMU. ZAPRASZAMY DO KONTAKTU Z NASZYM BIUREM OBSŁUGI KLIENTA TEL. 22 349 60 12 W CELU ZAPOZNANIA SIĘ Z PEŁNĄ OFERTĄ BADAŃ. |
1. Markery stresu i zaburzeń metabolizmu komórkowego
Stres oksydacyjny to zaburzenie homeostazy, prowadzące do zaburzenia równowagi prooksydacyjno – antyoksydacyjnej w kierunku reakcji utleniania. W wyniku tego dochodzi do dezintegracji i uszkodzenie komórek, co skutkuje zaburzeniem ich funkcjonowania.
Nasza oferta zawiera pakiet 3 badań diagnozujących stres oksydacyjny:
Istnieją teorię, że stres oksydacyjny może odgrywać ogromną rolę w powstawaniu zaburzeń autystycznych. Antyoksydanty mogą poprawiać obraz choroby u tych pacjentów. Ważna w takim przypadku jest suplementacja substancjami ochronnymi, do których należą: witamina C i E, beta-karotyna, koenzym Q10, selen, cynk, miedź, mangan, substancję roślinne z grup karotynidów, polifenoli, fitoestrogenów i sulfidów. Działają one jako wychwytywacze wolnych rodników, chelatują metale i chronią komórki przed utlenieniem.
Równie poważnym zaburzeniem co stres oksydacyjny jest stres azotowy polegający na nadmiernym tworzeniu tlenku azotu (NO) i produktów jego metabolizmu: peroksynitrytu, nitrotyrozyny i kwasu nitrofenylooctowego.
Tlenek azotu (NO) jest wolnym rodnikiem, który jest tworzony niemal we wszystkich komórkach ludzkiego organizmu. Z powodu swoich niewielkich rozmiarów i wysokiej lipofilności przenika on szybko przez bariery. Ma on krótki czas działania ale wysoką aktywność biologiczną. Z tlenem reaguje szybko przechodząc w nitraty.
Tworzenie tlenku azotu (NO) zachodzi poprzez enzym – syntetazę tlenkoazotową (NOS) z L-argininy. Jako produkt uboczny tej rekcji powstaje cytrulina i woda.
Tlenek azotu pełni ważne funkcje fizjologiczne. Syntetaza tlenkowęglowa występuje w czterech postaciach izomerycznych:
Z powodu wysokiego powinowactwa do grup hydroksylowych, peroksylowych, tyrozynowych NO jest wolnym rodnikiem. W fizjologicznych stężeniach hamuje on oksydację lipidów. Kolejnymi zadaniami fizjologicznym są hamowanie agregacji trombocytów i wzmaganie odpowiedzi na insulinę.
Cykl Krebsa stanowi centralny punkt przemiany materii. W nim odbywa się rozkład węglowodanów tłuszczy i białek do Ac-CoA. Substancja ta jest metabolizowana w celu pozyskania energii. Powstaje przy tym GTP oraz produkty reakcji: NADH i FADH , które są dalej przetwarzane w ATP. Cykl Krebsa jest także poprzez swoje produkty pośrednie punktem wyjścia do rozmaitych biosyntez. Temu służy np. Alfa-ketoglutaran jako substancja wyjściowa do tworzenia aminokwasów, glutaminy, argininy, proniny. Z bursztynylo-CoA jest wytwarzany kwas deltaaminolewuliunowy, który dalej jest substancją bazową do biosyntezy hemu.
Wysokie poziomy NO hamują enzym akonidazę i uniemożliwiają przemianę cytrynianu i izocytrynianu. Powoduje to blokadę całego cyklu Krebsa i uniemożliwia metabolizowanie białek , tłuszczy i aminokwasów. W tym mechanizmie dochodzi do niedoboru NADH i FADH2 i wynikający stąd niedobór substratów do syntezy ATP w mitochondriach. W konsekwencji w komórkach pojawia się deficyt energetyczny.
Dodatkowe przyspieszenia przemiany materii np. poprzez fizyczny, psychiczny stres, infekcje lub odżywanie bogate w węglowodany wzmagają wielokrotnie syntezę NO. Powstaje ekstremalnie wysoki deficyt energii w którym dochodzi także do blokady oksydacji kwasów tłuszczowych oraz blokady rozkładu węglowodanów (glikolizy). W procesie glikolizy z glukozy tworzony jest kwas pirogronowy oprócz tego jest on produktem ubocznym rozkładu aminokwasów: glicyny, cysteiny, alaniny. Przy wystarczającym poziomie energii w komórce, przy pomocy enzymu dehydrogenazy pirogronowej dochodzi do jego przemiany w Ac-CoA. W sytuacji stresu azotowego ten ważny szlak metaboliczny jest uszkodzony z powodu niedostatecznej aktywności enzymatycznej. Prowadzi to do osłabienia wydajności energetycznej komórek. Kwas pirogronowy zamiast przemiany w Ac-CoA jest redukowany do kwasu mlekowego. Mając na uwadze to zjawisko można mierzyć zaburzenia metabolizmu poprzez określanie stosunku kwasów: mlekowego do pirogronowego. Im wyższe wartości przyjmuje ten wskaźnik tym bardziej zaburzony jest bilans energetyczny.
Jakiekolwiek komórki produkujące cytokiny mogą stymulować NO. Nie wiadomo, czy nadmierna produkcja NO w autyzmie jest umiejscowiona w konkretnych organach czy tkankach. Najprawdopodobniej ma to miejsce w mózgu i przewodzie pokarmowym, oba te układy w autyzmie funkcjonują nieprawidłowo, w obrazie pojawiają się objawy behawioralne i gastrologiczne.
Nadmierny poziom NO w mózgu zwiększa apoptozę , uszkadza barierę krew-mózg, zwiększa neurodegenerację i demielinizację . Takie mechanizmy mogą mieć wpływ na obraz choroby w zespole autystycznym.
Zwiększenie produkcji NO zachodzi także poprzez ekspozycję na obce substancje: chemikalia, metale ciężkie, leki (antybiotyki, cytostatyki, statyny). Wielu autorów doszukuje się związku pomiędzy ekspozycją na metale ciężkie (szczególnie rtęć) oraz spożywaniem acetaminofenu (paracetamol) zarówno przez dzieci jak i przez matki w ciąży. Substancje te wywołują w organizmie stres azotowy.
Nadmierne tworzenie NO zaburza metabolizm. NO posiada silne powinowactwo do żelaza i siarczku żelaza co hamuje działanie enzymów. Duże ilości NO hamują cykl oddechowy mitochondriów. Spowodowana tym utrata ATP dotyka w szczególności komórki o wysokim zapotrzebowaniu energetycznym a więc komórki nerwowe i komórki układu immunologicznego. W rezultacie aktywują się receptory glutaminianowe w tkance nerwowej (synapsach). Glutaminian wypiera blokujący kanał jon magnezu i otwiera go powodując napływ jonów wapnia do komórki nerwowej. W stresie azotowym proces ten jest silnie wzbudzony. Powoduje to utratę funkcji komórki nerwowej i częściową blokadę przewodnictwa. Dłużej trwający napływ wapnia prowadzi do obumierania komórek. Zjawisko takie nazywa się ekscytotoksycznością.
Nadmiar wapnia uruchamia aktywację syntazy tlenku azotu (iNOS) i kinazę białkową C (PKC). iNOS powoduje produkowanie w nadmiarze tlenku azotu, który zaczyna kumulować się w komórkach. Gdy napotka wolne rodniki tworzy bardzo destrukcyjną cząsteczkę, która uszkadza mitochondria – główne źródło energii dla neuronu.
W tym samym czasie kinaza białkowa C aktywuje fosfolipazę A2 w membranie komórkowej, co sprawia że kwas arachidonowy jest wypuszczany do cytozolu. Tam działają na niego dwa enzymy: lipoksygenaza i cyklooksygenaza, które produkują potencjalnie destrukcyjne eikozanoidy. Najistotniejszym jest enzym COX II, który prowadzi do kumulacji prostaglandyn PGE2 i PGD2, które wzmagają stan zapalny.
Kumulacja eikozanoidów prowadzi do produkowania wolnych rodników, w szczególności bardzo destrukcyjne rodniki hydroksylowe. Proces ten przyspiesza i wolne rodniki wchodzą w interakcje z membranami neuronów, w tym membranami mitochondrialnymi i komórkowymi. Gdy ten proces się zacznie, początkuje on reakcję łańcuchową w wielonasyconych kwasach tłuszczowych, z których zrobione są membrany. Proces ten nazywamy peroksydacją lipidową i jest to objaw stresu oksydacyjnego. Jak widać zatem: obydwa rodzaje stresu: tlenowy i azotowy są ze sobą powiązane.
Aktywacja mikrogleju i astrocytów w procesie zapalnym, poprzez stymulację układu odpornościowego może wywołać ekscytotoksyczność. Aktywacja mikrogleju powoduje uwolnienie się licznych cytokin, w tym TNF-alfa, IL-1ß, IL-2, IL-6 i INF-gamma. Dodatkowo, ma miejsce aktywacja prozapalnych eikozanoidów. Powiązane z tym procesem jest wygenerowanie różnego rodzaju produktów pośrednich przemian tlenowych i azotowych, w tym nadtlenku wodoru, rodników wodorotlenowych, nadtlenoazotynu i 4-hydroksynonenalu. Te produkty pośrednie nie tylko uszkadzają neurony, połączenia synaptyczne i części składowe komórek ale indukują też uwolnienie glutaminianu z sąsiadujących astrocytów.
Aktywacja mikrogleju może również spowodować uwolnienie się glutaminianu i kwasu chinolinowego – dwóch silnych ekscytotoksyn – z samego makrofagu. Jest to związane z uruchomieniem alternatywnego szlaku metabolizmu tryptofanu. Wszystko, co prowadzi do aktywacji mikrożelu, w tym wirusy, ß-amyloidy, rtęć, aluminium, oksydowany LDL i HDL, homocysteina i ekscytotoksyny, może wpłynąć na kumulację kwasu chinolinowego. Szczególnie istotna jest równowaga między kwasem chinolinowym a kynurenitami, które pełnią dla mózgu funkcję ochronną.
Interakcje z bakteriami, wirusami i lipopolisacharydami mogą zwiększyć wydzielanie się glutaminianu 2-3 krotnie powyżej normalnego poziomu. Należy również zauważyć, że nadmiar glutaminianu, tak jak i jego niedobór, wpływa na długoterminowe wzmocnienie kluczowe dla procesów uczenia się i zapamiętywania. Dodatkowo, wzrost i kierunek rozwoju ścieżek w mózgu jest również regulowany przez glutaminian, w szczególności wpływ na niego ma przedłużający się okres nadmiaru glutaminianu. Niedobór glutaminianu zaburza funkcję stożka wzrostu i prowadzi do niewłaściwej budowy mózgu.
Podczas oksydacji lipidowej powstają produkty uboczne w tym kilkanaście różnych aldehydów. Najbardziej powszechny jest malondialdehyd (MDA), a najbardziej destrukcyjny jest 4-hydroksynonenal (4-HNE). Ostatnie badania dowiodły, że 4-HNE może w dużym stopniu niszczyć komórki, włącznie z zahamowaniem defosforylacji białka tau, co wpływa na funkcję mikrotubuli. Dowiedziono także, że hamuje on reduktazę glutationową, która jest niezbędna do przemiany utlenionego glutaminianu do jego funkcjonalnej, zredukowanej formy. Stwierdzono, że dzieci z chorobami autoimmunologicznymi mają wyższe poziomy 4-HNE we krwi niż dzieci z grupy kontrolnej.
Stwierdzono również, że 4-HNE może łączyć się z proteinami synaps, gdzie upośledza transport zarówno glukozy, jak i glutaminianu. Ten proces jest wyjątkowo niebezpieczny, bo liczne badania wykazały, że upośledzenie źródeł energii znacznie zwiększa podatność na działanie glutaminianu do tego stopnia, że nawet normalne poziomy glutaminianu mogą być ekscytotoksyczne.
Wiadomo już również z badań doświadczalnych, że napady drgawkowe są blisko związane z procesem ekscytotoksyczności . Nie tylko glutaminian i kwas aspartamowy mogą wywoływać drgawki, ale same drgawki mogą stymulować uwalnianie się ekscytotoksyn w mózgu, prawdopodobnie przez stymulację powstawania wolnych rodników. Spontanicznie niszczone neurony, w szczególności gdy proces ma miejsce przez długi czas, powodują utratę energii, niedokrwienie i niedotlenienie, które to powodują również nadmierne uwalnianie się glutaminianu.
Podczas gdy degeneracja neuronów może brać się z zbyt wysokich poziomów glutaminianu, utrata połączeń dendrytowych może mieć miejsca przy dużo mniejszych stężeniach. Wiadomo również, że niedotlenienie i niedokrwienie, częste przy drgawkach występujących w długim okresie czasu, mogą również drastycznie zwiększać poziom glutaminianu. Może mieć to duże znaczenie przy formowaniu się ścieżek, jak również wpływa na utratę neuronów, połączeń synaptycznych i komórek pnia mózgu. Wiadomo, że po osiągnięciu wieku 2 lat rozwijający się mózg zawiera więcej receptorów glutaminianu niż po urodzeniu i ta liczba powoli zmniejsza się przez następną dekadę. Dlatego mózgi dzieci są bardziej podatne na ekscytotoksyczność.
W stresie azotowym dochodzi do tworzenia się peroksynitrytu (ONOOˉ) – wskutek nadmiaru NO lub niedostatecznej aktywności manganozależnego enzymu dysmutazy nadtlenku. Pozostający w nadmiarze ONOOˉ ma duże powinowactwo do aminokwasów szczególnie do tyrozyny i tryptofanu. W połączeniu z tyrozyną tworzona jest nitrotyrozyna, która jest związkiem bardzo stabilnym i nieulegającym dalszemu metabolizmowi. Jest to zatem dobry wskaźnik stresu azotowego.
Badamy nitrotyrozynę metodą wysokosprawnej chromatografii cieczowej, która jest złotym standardem w oznaczaniu tego związku, daleko przewyższającym metody immunoenzymatyczne.
Kwas pirogronowy jest jednym z podstawowych substratów w cyklu Krebsa w tworzeniu Ac CoA. Jego zmniejszony poziom świadczy o niedostatecznym trawieniu węglowodanów, zwiększenie poziomu świadczy o niedostatecznej funkcji enzymu przekształcającego do Ac CoA – co ma bezpośredni wpływ na wydajność energetyczną komórek. W sytuacji tego bloku zamiast do Ac-CoA kwas pirogronowy redukowany jest do mleczanu. Wzrost wartości ilorazu kwas mlekowy/kwas pirogronowy jest markerem niewydolności energetycznej organizmu poprzez blokadę cyklu Krebsa. Oba parametry oznaczane są metoda spektrometrii masowej zapewniającej najwyższą możliwą dokładność pomiaru.
Wiele danych klinicznych i biochemicznych dowodzi, że podstawowy aminokwas - Tryptofan stanowi „Missing link” między schorzeniami somatycznymi i psychicznymi.
Tryptofan stanowi podstawowy aminokwas, metabolizowany w warunkach fizjologicznych w serotoninę i melatoninę. We krwi tryptofan wiąże się w 80 do 90 % z albuminą i w osoczu występuje jedynie niewielka jego ilość. Jedynie około 1 procent przyjętego tryptofanu jest przekształcany w serotoninę. Przy tym największy udział syntezy serotoniny ma miejsce w komórkach enterochromafinopodobnych przewodu pokarmowego, a tylko bardzo nieznaczna część w centralnym układzie nerwowym.
Krążący w organizmie tryptofan konkuruje o wchłonięcie do mózgu z innymi aminokwasami. Dostępność tryptofanu dla neuronów serotoninergicznych jest zależna od stosunku pomiędzy tryptofanem, a stężeniami tych konkurujących aminokwasów we krwi. Wzmożoną synteza serotoniny można zatem osiągnąć zarówno podnosząc poziom wolnego tryptofanu jak poprzez obniżenie poziomu aminokwasów konkurujących z tryptofanem o przeniknięcie do mózgu (np. spadek waliny, leucyny i izoleucyny po posiłku w wyniku zwiększonego dopływu węglowodanów – spowodowany przez stymulację wydzielania insuliny oraz od-insulinowe zwiększone wchłanianie przede wszystkim waliny, leucyny i izoleucyny przez mięśnie, por. Hüther et al., 2000).
W warunkach fizjologicznych prawie jeden procent tryptofanu zostaje przetransferowany przez enzym tryptofan-5-hydroksylazę (TPH) do 5-hydroksytryptofan (5-http). Ta ścieżka metabolizmu potrzebuje jako kofaktorów kwasu foliowego, witaminy B3, żelaza, miedzi i witaminy C. Hiperinsulinemia, hiperkortyzolizm (stres) i niedobór witamin B3 oraz B6 prowadzą do blokady enzymów. Powstający w ramach stresu azotowego azotyn nadtlenowy również blokuje hydroksylazę tryptofanu. 5-http jest następnie transferowany w serotoninę przez aromatyczną karboksylazę aminokwasu L. Witamina B6 działa przy tym jako kofaktor.
Rozpad tryptofanu ma miejsce w sposób fizjologiczny dzięki wątrobowej 2,3-dioksygenazie tryptofanowej (TDO) za pośrednictwem ścieżki metabolizmu kinureniny (patrz ilustracja 2). 2,3-dioksygenaza tryptofanowa jest indukowana przez tryptofan, kinureninę i hormony steroidowe jak kortyzol i estrogeny, zaś hamowana przez NADPH.
Kwas 5-hydroksyindolooctowy (5-HIAA) stanowi główny metabolit serotoniny. Podwyższone stężenia występują w przypadku karcynoidów (guzów neuroendokrynnych) lub w leczeniu za pomocą inhibitorów wychwytu zwrotnego serotoniny (SSRI – Selective Serotonin Reuptake Inhibitor).
Ilustracja 1 ukazuje, że do początkowej hydroksylacji tryptofanu jest potrzebna jako kofaktor tetrahydrobiopteryna (BH4). Zredukowane poziomy BH4 mogą być uwarunkowane przez mutacje genetyczne w enzymach uczestniczących w syntezie-regeneracji BH4, a to ze względu na niedobór kwasu foliowego, zwiększonego stresu oksydacyjnego oraz podczas metabolicznych stanów zapalnych.
Pacjenci z obniżonym statusem BH4 wykazują progresywne opóźnienie psychomotoryczne, dystonię oraz ciężki niedobór dopaminy i serotoniny z obniżonym poziomem 5-HIAA oraz kwasu homowanilinowego (Bonafe et al., 2001). Homowanilina stanowi najważniejszy metabolit dopaminy.
Tryptofan
Fe, wit. B3, BH 4, hydroksylaza tryptofanowa (TPH)
⇩
5-hydroksy-tryptofan
wit. B6, aromatyczna karboksylaza aminokwasu L.
⇩
Serotonina ⇨ kwas 5-hydroksyindolooctowy
⇩
N-acetyloserotonina
⇩
Melatonina
Ilustr. 1: metabolizm tryptofanu-serotoniny
| 5-HIAA ⇧ (podwyższony poziom5-hydroksy-tryptofanu) | 5-HIAA ⇩ (obniżony poziom5-hydroksy-tryptofanu) |
|---|---|
|
|
Wytwarzanie kwasu ksanturenowego ma miejsce w trakcie metabolizmu tryptofanu za pomocą metabolizmu kinureniny. Niedobór witaminy B6 prowadzi do spowolnienia ściśle zależnego od witaminy B6 postępu syntezy ze skutkiem akumulacji hydroksykinureniny i kinureniny (Chiang et al., 2005; Tada et al., 1968; Kosters et al., 1976). Oba te metabolity są szybko metabolizowane dalej w kwas ksanturenowy oraz kwas kinureninowy i pojawiają się w podwyższonych ilościach w moczu (patrz ilustr. 2; Knapp, 1961; Abbassy 1959). Podwyższone poziomy kwasu ksanturenowego świadczą o niedoborze witaminy B6, zwłaszcza wtedy, gdy wydalanie kwasu chinolinowego w moczu nie jest podwyższone. Kwas kinureninowy jest wydalany w niewielkich ilościach, ponieważ jest ona dalej metabolizowany w cyklu kwasu cytrynowego (Matte et al., 2001).
Podwyższone poziomy kwasu ksanturenowego mogą stanowić skutek niedoboru witaminy B6, modulacji metabolizmu kinureniny przez hormony steroidowe oraz skutek zredukowanego wskaźnika rotacji tryptofanu w wyniku zmniejszonego dostarczania protein.
Podwyższone poziomy kwasu ksanturenowego indukują wolne rodniki. Hydrolizowaną strukturę choinową kwasu ksanturenowego wiąże żelazo i tworzy w ten sposób kompleks uszkadzający oksydacyjnie DNA (Murakami et al. 2006).
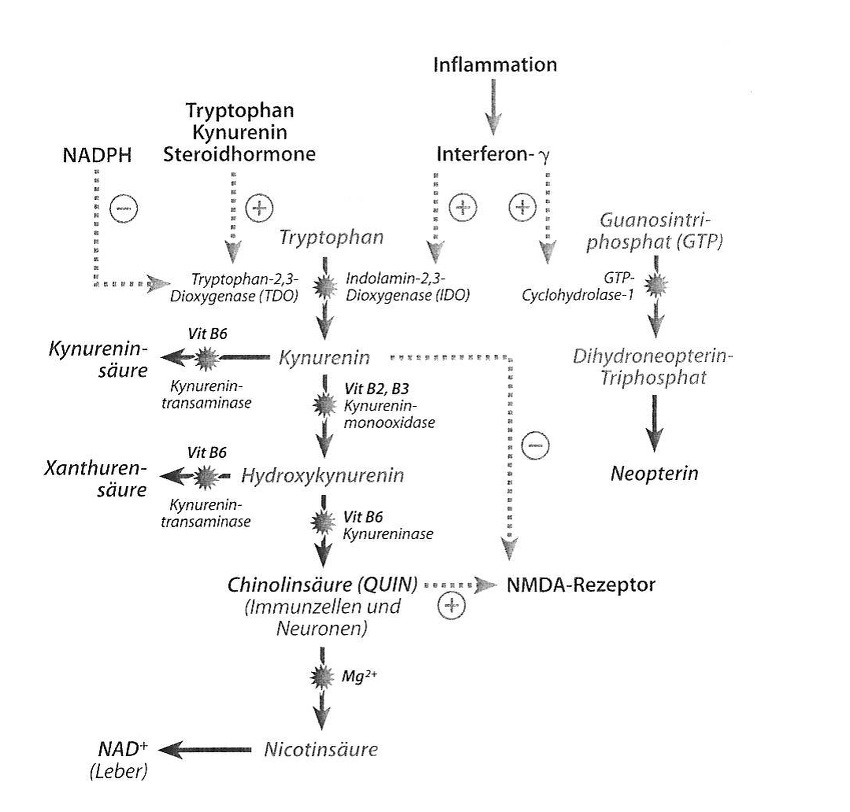
Ilustr. 2: Rozpad tryptofanu ma miejsce w warunkach fizjologicznych oksydacyjnie dzięki 2,3-dioksygenazie tryptofanowej (TDO). W zależności od tkanki powstaje przy tym produkt końcowy NAD+ (wątroba) lub kwas chinolinowy (komórki odpornościowe i neurony). W okolicznościach patologicznych (chorobowych) jak np. stan zapalny, zostaje zaindukowana 2,3-dioksygenaza indolaminowa (IDO), co prowadzi do wzmożonego wytwarzania kwasu chinolinowego. Jeśli aktywność enzymów w wyniku braku kofaktorów (witamina B2, B3, B6) zostanie obniżona lub też TDO zostanie wzmocnione w swej aktywności przez hormony steroidowe lub wzmożony metabolizm tryptofanu, to dochodzi do wzmożonej syntezy kwasu kinureninowego oraz ksanturenowego.
Szereg mechanizmów immunologicznych i endokrynologicznych może prowadzić do oksydacyjnej indukcji metabolizmu tryptofanu (patrz ilustr. 2). Przekształcenie tryptofanu w kinureninę jest katalizowane przez dwa enzymy: przez usytuowaną w wątrobie 2,3-dioksygenazę tryptofanową (TDO) oraz przez znajdującą się przede wszystkim w makrofagach, astrocytach i w mikroglejach 2,3-dioksygenazę indolaminową (IDO). IDO jest indukowana przez prozapalne cytokiny jak interferon-γ. Mechanizmy prozapalne jak aktywowanie podstawowych czynników jądrowych (NF-KB) stres azotowy stanowią przy tym również istotne mechanizmy patologiczne (Alberti-Giani et al., 1997).
Inne cytokiny jak TNF-α i interferon-α ingerują w sposób regulujący w stopień aktywacji oraz okres trwania aktywacji ścieżki kinureninowej.
W wyniku indukcji enzymu zostaje uruchomiony wzmożony katabolizm tryptofanu ukierunkowany na metabolizm kinureniny. Wynikiem jest spadek stężenia tryptofanu w osoczu ze zredukowaną zdolnością biosyntezy serotoniny, która objawia się w obszarze przewodu pokarmowego przez zaburzenia ruchów robaczkowych oraz bolesności, zaś w obszarze centralnego układu nerwowego przez depresje i stany lękowe. Poza tym aktywacja IDO prowadzi do zwiększonej produkcji kwasu chinolinowego (QUIN) – silnego agonisty receptora NMDA (patrz ilustr. 2).
W makrofagach i mikroglejach (makrofagi centralnego układu nerwowego (ZNS)) powstaje w wyniku indukcji metabolizmu kinureniny szereg neuroaktywnych substancji, jak np. QUIN oraz hydroksykinurenina. QUIN stanowi krytyczny link między układem odpornościowym a ZNS. QUIN jest agonistą na receptorze glutaminianu typu NMDA. Aktywacja prowadzi do odczuwania bólu i stanowi klasyczny mechanizm biochemiczny typowej objawowości bólowej w infekcjach wirusowych. Jeśli receptor NMDA jest przesadnie stymulowany, prowadzi to do wewnątrzkomórkowego napływu wapnia do neuronów glutaminergicznych z degeneracją neuronów oraz znaną egzotoksycznością glutaminianową (Molz et al., 2007). Z tego patomechanizmu wywodzi się znaczenie QUIN w różnych schorzeniach neuro-zapalnych, jak np. choroba Alzheimera. Wykazano, że QUIN indukuje ekspresję IL-1 w astrozytach. IL-1 stanowi kluczowego mediatora w patogenezie choroby Alzheimera (Ting et al., 2009).
Podczas gdy QUIN stanowi agonistę na receptorze glutaminianu typu NMDA, to kinurenina działa sama jako antagonista na receptorze NMDA (patrz ilustr. 2). Oznacza to, że QUIN działa jak neurotoksyna, podczas gdy sama kinurenina wywiera skutek wspierający neurony. Trzeci metabolit – hydroksykinurenina – może już w niewielkich dawkach prowadzić do indukcji wolnych rodników a tym samym do stresu oksydacyjnego i azotowego z następstwami jakie stanowi degeneracja neuronów (Stone, 2003).
QUIN działa jak agonista NMDA czyli neurotoksyna, podczas gdy kinurenina ma działanie protekcyjne wobec neuronów jako antagonista NMDA. Hydroksykinurenina działa neurodegeneracyjnie w wyniku indukcji stresu oksydacyjnego i azotowego.
Toksyny, jak metylortęć mogą podwyższać aktywność QUIN w receptorze NMDA. W wyniku doświadczeń na zwierzętach (szczury) wykazano, że narażenie na metylortęć prowadzi do znacznej aktywacji ścieżki kinureniny i upośledza rozwój mózgu (Zanoli et al. 2001). Ftalany, stosowane na całym świecie w wyrobach plastikowych wykazują również zdolność do zwiększania wytwarzania QUIN w wyniku blokady fizjologicznego metabolizmu tryptofanu (Fukuwatari et al., 2004).
W badaniu analizowany jest poziom metabolitów tryptofanu w moczu. Tryptofan jest aminokwasem będącym substratem do produkcji serotoniniy i melatoniny, ma wpływ na funkcje wątroby, a także bierze udział w syntezie koenzymu NAD. Podwyższony poziom tryptofanu w moczu obserwujemy w zaburzeniach przemiany materii, które powodują niedobór witaminy B6 i niacyny. Zwiększone wydalanie tego związku obserwujemy także w przypadkach zwiększonej substytucji. Zaburzenia transportu aminokwasów i funkcji nerek również mogą wpłynąć na podwyższenie tego parametru w moczu.
W próbce oznaczane są;
Obliczany jest także stosunek L-kinurenina/tryptofan, a także stosunek kwasu kinureninowego/L-kinureniny.
System karnitynowy umożliwia transport aktywnych metabolicznie kwasów tłuszczowych poprzez błonę mitochondrialną do miejsc beta-oksydacji w formie karnityzowanych kwasów tłuszczowych i tym samym stanowi ważny element systemu energetycznego komórki. W procesie tym ważne są 4 enzymy:
Niskie wartości acetylo-L-karnityny wskazują pośrednio na zmniejszona aktywność acetylotransferazy karnitynowej, jak i zmniejszony poziom L-karnityny zgodnie z wzorem:
L-karnityna + AcCoA -> Acetyl-L-karnityna + CoA
Niedobory L-Karnityny i Acetyl-L-Karnityny prowadzą do zmniejszonej zdolności komórek do generowania energii pochodzącej z utleniania kwasów tłuszczowych i tym samym do zmniejszenia tworzenia Acetylokoenzymu A i zależnych od niego procesów metabolicznych. Kwasy tłuszczowe mogą przy tym ulegać przemianom w cholesterol lub inne glicerydy co prowadzić może np. do tworzenia blaszek miażdżycowych.
Glutation (GSH) jest peptydem mającym silne zdolności antyoksydacyjne. Uczestniczy on w wielu procesach ochronnych. Oznaczenie całkowitego glutationu w odniesieniu do hemoglobiny daje pogląd na zdolności samoobronne organizmu. Przy wielu chorobach przebiegających ze stresem azotowym i tlenowych pojawia się on w formie oksydowanej (GSSG). Określenie stosunku GSH/GSSG daje pogląd na przebieg procesów antyoksydacyjnych.
2. Diagnostyka zaburzeń jelitowych
Badanie umożliwia przeglądową ocenę stanu jelita. Analizowane są w nim następujące składowe:
Dysbioza to stan zdrowia, w którym doszło do zaburzenia normalnej flory bakteryjnej i grzybiczej zamieszkującej jelita. Czynnikami, które mogą wpłynąć na ten stan są m.in. zmiana dietetyczna, zażywanie antybiotyków i aktywność układu odpornościowego.
Objawy związane z tym stanem:
| Przerost drożdżaków/grzybów: | Przerost nieprawidłowych bakterii: |
|
|
Osobami, które są najbardziej podatne na dysbioze są:
Pośredni test dysbiozy wykonuje się w celu oceny składu flory jelitowej. Badanie to wykonywane jest z moczu na zawartość kwasów organicznych będących metabolitami bakterii zasiedlających przewód pokarmowy. Ocena ilości tych związków w badanym materiale może być źródłem istotnych informacji na temat nieprawidłowej flory bakteryjnej zasiedlającej jelita m.in.:
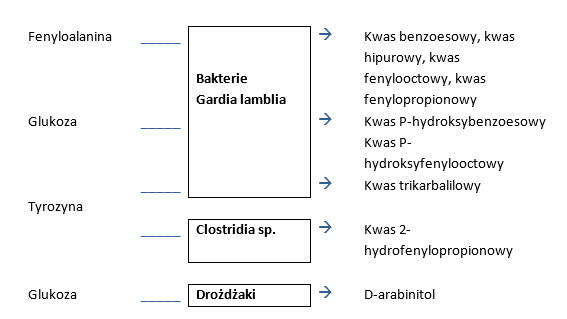
Korzyści diagnostyczne w wyniku zastosowania Pośredniego testu dysbiozy
Zonulina jest białkiem biorącym udział w regulacji przepuszczalności szczelin międzykomórkowych nabłonka jelita. Przyłączanie zonuliny do enterocytów aktywuje procesy biochemiczne doprowadzające do otwierania ścisłych połaczeń (tzw. tightjunctions) i skutkuje wzmożoną przepuszczalnością jelit. Podwyższony poziom zonuliny często występuje u osób cierpiących na choroby takie jak celiaklia, cukrzyca typu I, reumatoidalne zapalenie stawów czy stwardnienie rozsiane.
Histamina odgrywa ważną rolę w procesach zapalnych i reakcjach alergicznych.
Nietolerancja histaminy polega na wzmożonej reakcji na histaminę.
Nadmiar histaminy w organizmie może być spowodowany przez:
W zależności od stężenia histaminy dochodzi do miejscowych stanów zapalnych błony śluzowej jelit bądź po wchłonięciu – do reakcji ogólnoustrojowych. Obserwuje się wtedy szereg objawów ze strony układu pokarmowego: zgagę, mdłości, biegunkę, skurcze jelit. Histaminoza ogólnoustrojowa może manifestować się w postaci rumienia, bólu głowy, tachykardii, niedociśnienia, trudnościami w oddychaniu czy bólami skurczowymi jelit.
W moczu oznaczana jest histamina, histydyna (prekursor histaminy), imidazol-4-yl-kwas octowy, N-metylohistamina i N-metylimidazol-4-yl-kwas octowy. Wszystkie wyniki podane są w przeliczeniu na gram kreatyniny. W stanie zdrowia w moczu obserwujemy bardzo małe ilości histaminy. Jej wzrost może świadczyć o reakcjach alergicznych lub chorobach wywołujących wzrost komórek tucznych pomimo braku reakcji alergicznych.
Z nietolerancją histaminy mamy do czynienia, jeśli na spożytą wraz z pokarmem histaminę organizm reaguje objawami nietolerancji. Może to mieć miejsce, gdy w organizmie znajduje się zbyt mała ilość degradującego histaminę enzymu diaminooksydazy (DAO) lub też jeśli do organizmu dostało się więcej histaminy, niż jest on w stanie zdegradować. U osób zdrowych pożywienie zawierające histaminę jest degradowane już w jelicie, przy czym szybkość degradowania jest określana przez aktywność diaminooksydazy. U pacjentów z objawami nietolerancji histaminy czynność DAO może być zredukowana.
Badanie histaminy w kale może służyć w diagnozowaniu nietolerancji pokarmowej, która może być wywołana histaminą. W zależności od stężenia histaminy dochodzi do miejscowych stanów zapalnych błony śluzowej jelit bądź po wchłonięciu – do reakcji ogólnoustrojowych. Obserwuje się wtedy szereg objawów ze strony układu pokarmowego: zgagę, mdłości, biegunkę, skurcze jelit. Histaminoza ogólnoustrojowa może manifestować się w postaci rumienia, bólu głowy, tachykardii, niedociśnienia, trudnościami w oddychaniu czy bólami skurczowymi jelit.
Bardzo ważne w przypadku tego badania jest dokładne zanotowanie wszystkich produktów zawierających histaminę, które zostały spożyte w dniu poprzedzającym pobranie kału do badania.
EPX jest cytotoksycznym białkiem wydzielanym przez zaktywowane eozynofile w reakcji obronnej przed patogenami takimi jak bakterie i pasożyty. Stanowi marker do rozpoznania i oceny reakcji zapalanych w jelicie. Niewielkie podwyższenie poziomu EPX w kale sugeruje utajony stan podrażnienia przez antygeny (składniki pokarmowe, mikroorganizmy). W przypadku znacznego zwiększenia poziomu EPX będącego wyrazem nasilonych reakcji immunologicznych, podejrzewa się obecność pasożytów jelitowych. Po za tym duża ilość EPX wskazuje na alergie i nadwrażliwości pokarmowe typu IgG4.
Defensyny stanowią pierwszą linię obrony przeciwko mikroorganizmom bytującym na skórze i błonach śluzowych. Są ważnym elementem układu immunologicznego, gdyż zwalczają szerokie spektrum mikroorganizmów chorobotwórczych i toksyn, znajdujących się na błonie śluzowej jelita. Defensyny są wydzielane przez neutrofile i komórki nabłonkowe błony śluzowej jelit. Obniżony poziom β-defensyny w kale świadczy o słabszym działaniu obronnym śluzówki jelit oraz zmniejszoną neutralizacją antygenów. Z kolei podwyższone stężenie wskazuje na wzmożone reakcje obronne przeciwko mikroorganizmom, toksynom, antygenom.
Nadwrażliwość pokarmowa IgG-zależna (NP-IGG) dotyczy reakcji pokarmowej z udziałem przeciwciał czterech podklas: IgG1, IgG2, IgG3 i IgG4. Występowanie NP-IGG wiąże się z występowaniem różnych symptomów, m.in. zaburzenia czynności jelit, nieswoiste zapalenie jelit, choroby stawów i układu oddechowego, zaburzenia dermatologiczne. Wymienione objawy chorobowe występują najczęściej w odległym czasie od spożycia alergenu pokarmowego. Rozpoznanie nadwrażliwości pokarmowej polega na oznaczaniu pokarmowo-swoistych przeciwciał podklas IgG, które są charakterystyczne dla poszczególnych produktów spożywczych.
Badania FoodProfil nie posiadają diagnostyki wstępnej. Wyniki jaki otrzymamy w teście to wyniki ilościowe, dla łatwiejszej interpretacji przedstawione są one w skali 0-3. Kolor zielony oznacza, że wyniki są istotne klinicznie, kolor żółty – wzmożoną reaktywność, a kolor czerwony – reaktywność wysoką.
Test FoodProfil 45 obejmuje badanie następujących składników pokarmowych:
Nadwrażliwość pokarmowa IgG-zależna (NP-IGG) dotyczy reakcji pokarmowej z udziałem przeciwciał czterech podklas: IgG1, IgG2, IgG3 i IgG4. Występowanie NP-IGG wiąże się z występowaniem różnych symptomów, m.in. zaburzenia czynności jelit, nieswoiste zapalenie jelit, choroby stawów i układu oddechowego, zaburzenia dermatologiczne. Wymienione objawy chorobowe występują najczęściej w odległym czasie od spożycia alergenu pokarmowego. Rozpoznanie nadwrażliwości pokarmowej polega na oznaczaniu pokarmowo-swoistych przeciwciał podklas IgG, które są charakterystyczne dla poszczególnych produktów spożywczych.
Te pakiety badań są prowadzone dwuetapowo. Wyniki ilościowe są przyporządkowane do klas 0-6. Kolor zielony oznacza, że wyniki są istotne klinicznie, kolor żółty – wzmożoną reaktywność, a kolor czerwony – reaktywność wysoką.
FoodProfil 88 obejmuje badanie następujących składników pokarmowych:
Nadwrażliwość pokarmowa IgG-zależna (NP-IGG) dotyczy reakcji pokarmowej z udziałem przeciwciał czterech podklas: IgG1, IgG2, IgG3 i IgG4. Występowanie NP-IGG wiąże się z występowaniem różnych symptomów, m.in. zaburzenia czynności jelit, nieswoiste zapalenie jelit, choroby stawów i układu oddechowego, zaburzenia dermatologiczne. Wymienione objawy chorobowe występują najczęściej w odległym czasie od spożycia alergenu pokarmowego. Rozpoznanie nadwrażliwości pokarmowej polega na oznaczaniu pokarmowo-swoistych przeciwciał podklas IgG, które są charakterystyczne dla poszczególnych produktów spożywczych.
Te pakiety badań są prowadzone dwuetapowo. Wyniki ilościowe są przyporządkowane do klas 0-6. Kolor zielony oznacza, że wyniki są istotne klinicznie, kolor żółty – wzmożoną reaktywność, a kolor czerwony – reaktywność wysoką.
FoodProfil 280 obejmuje badanie następujących składników pokarmowych:
Badanie to ma na celu różnicowanie pomiędzy rodzajami infekcji drożdżakami z rodzaju Candida. Drobnoustrój ten ma zdolności wypełniania najdrobniejszych luk w koloniach jelitowych i wrastania z błonę śluzową jelita. W początkowej fazie kolonizacji dochodzi do kontaktu z rozpuszczalnymi antygenami Candida przez co system immunologiczny reaguje odpowiedzią humoralną przy czym aktywowane są limfocyty Th2 odpowiedzialne za ten typ reakcji. W fazie przetrwałej kolonizacji uaktywniana jest odpowiedź komórkowa i aktywne są limfocyty Th1.
Test oparty jest o badanie obecności cytokin charakterystycznych dla obydwu typów limfocytów. Określane jest uwalnianie podstawowe IFN-γ i IL-10 oraz po stymulacji antygenem Candida. Przewaga wzrostu w jednym typie cytokin świadczy o nasileniu reakcji z udziałem odpowiedniego typu Th – Th1 – w przypadku IFN-γ i Th2 w przypadku IL-10.
Candida albicans jest grzybem z rzędu drożdżaków. Fizjologicznie może on wchodzić w skład flory jelitowej u większej części populacji. Wywołuje on najczęściej zakażenia u osób z obniżoną odpornością, stosujących przewlekle antybiotykoterapię, posiadających sztuczne zastawki, cewniki i po zabiegach inwazyjnych. Na rozwinięcie się zakażenia Candida może wpłynąć także nieprawidłowa dieta, brak snu, alkohol i nikotyna, stres i brak ruchu. Zakażenie tym drożdżakiem może rozwijać się zarówno na powierzchni jak i wewnątrz ciała – te rozwijające się wewnątrz, ogólnoustrojowe są zdecydowanie bardziej niebezpieczne dla organizmu.
3. Aktywność immunologiczna
Cytokiny to białka, które są wytwarzane przez komórki immunologiczne. Ich główną funkcją jest przekazywanie i regulacja odpowiedzi immunologicznej. Cytokiny działają poprzez przyłączanie się do swoistych receptorów na innych typach komórek, niż te przez które są wytwarzane. Mogą być one wykorzystane do stymulacji szpiku kostnego do wytwarzania komórek krwi u osób z supresją, jako czynniki przeciwnowotworowe i przeciwzapalne. W diagnostyce zostały również wykorzystane jako markery stopnia zaawansowania stanów chorobowych ich progresji, a także do oceny skuteczności leczenia.
4. Mikroelementy i witaminy, badania wykonywane z krwi pełnej zwiń opis
Przy obniżeniu poziomu witamin i mikroelementów obserwujemy wiele niespecyficznych objawów. Często ten deficyt przebiega bezobjawowo, jednak już przy niewielkich niedoborach zostają zaburzone funkcje enzymatyczne i immunologiczne. Określenie poziomu mikroelementów stanowi element diagnostyki pozwalający w pełni ocenić skale dolegliwości i deficytów oraz wdrożyć leczenie substytucyjne.
W skład tego badania wchodzą oznaczenia kwasu foliowego, witaminy B6 i witaminy B12 metodą wysokosprawnej chromatografii cieczowej.
Witamina B12 inaczej kobalamina jest produktem pochodzenia wyłącznie zwierzęcego. Jej funkcja w organizmie jest ściśle związana z działaniem kwasu foliowego biorącego udział w syntezie DNA. Bierze również udział w przemianie homocysteiny do metioniny, kwasu glutaminowego do kwasu β-metyloasparaginowego, a także kwasu metylomalonowego do bursztynowego.
Materiałem do tego badania jest mocz, w którym oznacza się:
Wszystkie metabolity są podawane w µg/g kreatyniny.
Badanie metabolitów witamin umożliwia ocenę niedoborów witamin z grupy B w organizmie. Nieprawidłowe ilości metabolitów mogą świadczyć o niedoborze, bądź zaburzeniu w metabolizmie tych witamin. Objawy jakie mogą temu towarzyszyć to m.in. niedokrwistość, zaburzenia ze strony układu nerwowego, problemy ze skórą, czy zmiany zapalne przewodu pokarmowego.
Specjalny zestaw do pobrania moczu porannego niestabilizowanego (ewentualnie można pobrać do zwykłego pojemnika na mocz – ilość moczu ok. 8,5 ml)
5. Badanie genetyczne
Gen MTHFR koduje enzym - reduktazę metylenotetrahydrofolianu. Białko to bierze udział w procesie przekształcenia aminokwasu homocysteiny do metioniny w reakcji katalizowanej przez syntazę metioninową, czyli odpowiada za usuwanie nadmiaru homocysteiny z organizmu. W genie MTHFR opisano kilkanaście polimorfizmów sekwencji DNA, które mogą spowodować spadek aktywności enzymu, co z kolei powoduje wzrost stężenia homocysteiny w surowicy krwi. Ma to negatywny wpływ na śródbłonek naczyń krwionośnych, powodując m.in. przerost mięśniówki naczyń, rozwój miażdżycy oraz zakrzepicy tętniczej i żylnej (szczególnie w obecności mutacji w genach czynnika II i V krzepnięcia krwi). Opisywany jest także możliwy związek pomiędzy obecnością polimorfizmów w genie MTHFR, a poronieniem nawracającym (szczególnie w I trymestrze ciąży), ostrą niewydolnością mięśnia sercowego, nefropatią cukrzycową, oraz autyzmem. Uważa się, że niedobór folianów w organizmie i obniżona aktywność MTHFR u kobiet mogą także zwiększać ryzyko powstania wrodzonych wad cewy nerwowej u potomstwa. W takim przypadku, gdy wykryje się u pacjentki wariantu genu wpływającego na zwiększone stężenie homocysteiny, zalecane jest badanie jej stężenia we krwi oraz badanie stężenia kwasu foliowego. Zaleca się suplementację kwasem foliowym oraz określonymi witaminami z grupy B ze względu na ich udział w metabolizmie homocyteiny.
Najczęściej występującym polimorfizmem w genie MTHFR jest zamiana cytozyny w pozycji 677 na tyminę (677C/T). Zmiana ta powoduje wbudowanie waliny w pozycji 222 w miejsce alaniny (A222V) w enzymie, czego wynikiem jest powstanie termolabilnego wariantu białka MTHFR o zmniejszonej aktywności. Homozygotyczny wariant 677 TT charakteryzuje się ok. 30-procentową aktywnością w porównaniu do typu dzikiego (CC) (homozygota TT występuje u ok.10% w populacji rasy kaukaskiej), podczas gdy heterozygoty (CT – ok. 40% osób rasy kaukaskiej) charakteryzują się około 60% aktywnością enzymu. Drugim często opisywanym polimorfizmem jest zmiana w pozycji 1298A/C i powoduje wymianę glutaminianu na alaninę w pozycji 429 (E429A) białka. Zamiana 1298A/C występuje w domenie regulatorowej genu MTHFR, w której to ma miejsce wiązanie S-adenozylometioniny (SAM). Mutacja 1298A/C powoduje obniżenie aktywności MTHFR, szczególnie u homozygot (ok. 10% osób rasy kaukaskiej, heterozygoty to ok. 40% osób rasy kaukaskiej) . Uważa się, że polimorfizm w pozycji 677 ma silniejszy efekt niż mutacja w pozycji 1298. Mutacja1298A/C ma mniejszy wpływ na stężenie homocysteiny w surowicy krwi, jednak w towarzystwie allelu 677T wzmacnia jego efekt i dodatkowo podwyższa stężenie homocysteiny, szczególnie u „podwójnych” homozygot i heterozygot.
6. Nagalaza zwiń opis
7. Endotoksemia
Wzmożone przedostawanie się bakterii lub bakteryjnych produktów przemiany materii do krwioobiegu jest określany jako translokacja bakteryjna i charakteryzuje się występowaniem podwyższonych stężeń endotoksyn we krwi (endotoksemia). Stałe obciążenie endotoksynami, pochodzącymi z jelita lub z płytki nazębnej może wywoływać w organizmie przewlekłe procesy zapalne niskiego stopnia (silent inflammation). Na skutek tego mogą one prowadzić do ciężkich schorzeń metabolicznych jak otyłość czy cukrzyca, lecz również do schorzeń układu sercowo-naczyniowego (miażdżyca naczyń, zawał serca, udar mózgu).
Endotoksyna, określana ze względu na swoją strukturę cząsteczkową również synonimem lipo-polisacharyd (LPS) jest odpornym na wysoką temperaturę komponentem zewnętrznej ścianki bakterii gram-ujemnych, który po obumarciu komórek bakterii ulega uwolnieniu. Endotoksyna składa się z odpowiedzialnej za reaktywność immunologiczną cząsteczkowej struktury rdzenia, stosunkowo jednolitego lipofilnego lipidu A oraz ze specyficznych pod względem gatunkowych, rozmaitych hydrofilnych, polisacharydowych łańcuchów bocznych. LPS działa jak silny aktywator komórek wrodzonego układu immunologicznego i w wyniku wyzwolonego przez tę aktywację wydzielania prozapalnych substancji posłańców wywołuje reakcje zapalne. Mogą one w rezultacie odpowiadać za ciężkie schorzenia metaboliczne lub sercowo-naczyniowe.
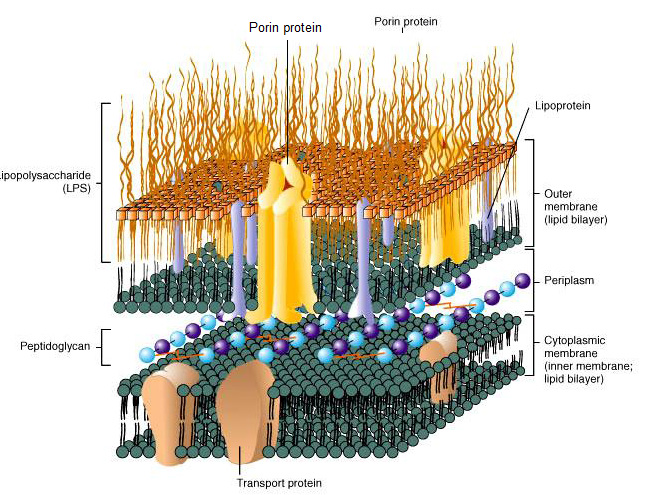
Ilustr. 1: Endotoksyna (LPS) stanowi integralny komponent ścianki komórki bakterii gram-ujemnych.
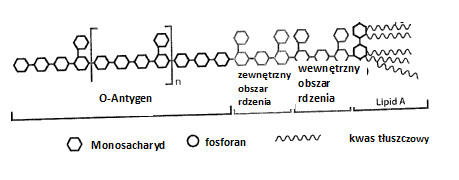
Ilustr. 2: Cząsteczkowa struktura endotoksyny (LPS)
Fagocyty jednojądrzaste układu immunologicznego (monocyty, makrofagi) są aktywowane przez wolną lub wiążącą się z LPS proteinę (LBP) skompleksowaną endotoksynę. Stymulacja przez przyłączony do receptora endotoksyny LPS inicjuje wewnątrzkomórkowy łańcuch sygnałów, którego skutkiem jest aktywacja i translokacja czynnika transkrypcji NF-K B do jądra komórki, gdzie odpowiada on po przyłączeniu się do regulatorowych regionów DNA za ekspresję genu różnych pośredników zapalenia. W rezultacie aktywowane w ten sposób komórki jednojądrzaste wytwarzają np. duże ilości prozapalnych cytokin (TNF-α, IL-1β, IL-6) mogących wywołać miejscowe lub ogólnoustrojowe reakcje zapalne. Dalszą konsekwencją aktywacji genu za pośrednictwem NF-KB jest ekspresja dającej się zaindukować syntezy tlenku azotu (iNOS). Ten enzym katalizuje wytwarzanie tlenku azotu (NO) z aminokwasu argininy i w ten sposób inicjuje wytwarzanie reaktywnych rodników azotu, które w przypadku ich nadprodukcji mogą stanowić wyzwalacz stresu azotowego i mitochondriopatii.
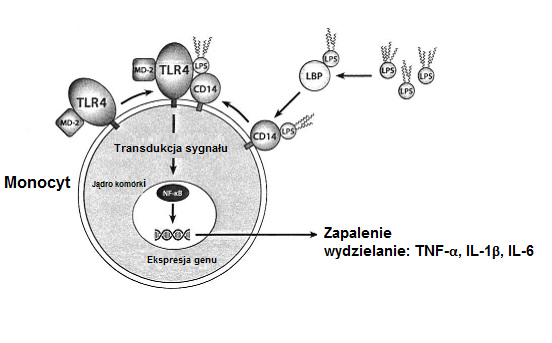
Ilustr. 3: Aktywowanie komórek odpornościowych przez endotoksynę (LPS)
Przewód pokarmowy stanowi naturalne schronienie i siedlisko dla stale obecnej w nim florze mikroorganizmów, składającej się zarówno z bakterii gram-dodatnich jak gram-ujemnych, które tworzą stabilny ekosystem. Pomimo licznego zasiedlenia bakteriami światła jelita organizm musi zagwarantować, że sterylność graniczących z nim jam i narządów zostanie zachowana i w związku z tym translokacja potencjalnie patogennych czynników chorobotwórczych zostanie udaremniona. W warunkach fizjologicznych śluz, nabłonek błony śluzowej oraz lamina propria nienaruszonych ścianek jelita, tworzą skuteczną barierę przeciwko przenikaniu bakterii lub ich komponentów, w wyniku czego endotoksyna, pochodząca od bakterii gram-ujemnych, dostaje się do krwi jedynie w niewielkich ilościach. Zmiana równowagi naturalnej jelitowej flory bakteryjnej na korzyść bakterii gram-ujemnych lub też zaburzenie funkcji bariery śluzówki jelitowej, prowadzące do podwyższonej przepuszczalności jelita może skutkować wzrostem translokacji endotoksyn do krwioobiegu.
Patologiczne znaczenie ogólnoustrojowego obciążenia organizmu mikroorganizmami przyzębia z jamy ustnej dla powstawania przewlekłych chorób zapalnych wyraźnie widać na przykładzie asocjacji zapalenia przyzębia ze zwiększonym ryzykiem występowania schorzeń sercowo-naczyniowych jak zawał serca czy udar.
Swobodna endotoksyna we krwi jest skompleksowana przez lipoproteiny, dzięki czemu biologiczna aktywność endotoksyny zostaje zneutralizowana. Głównym narządem do eliminacji endotoksyn z krwi jest wątroba. Właśnie w wątrobie LPS jest wchłaniany i metabolizowany przez komórki Kupffera – osiadłe makrofagi tkanki wątrobowej – a następnie oddawany do hepatocytów i wydalany za pośrednictwem żółci i jelita. Pochodzące z naturalnej flory bakteryjnej i przenikające do krwioobiegu niewielkie ilości LPS nie mają tym samym z reguły żadnego istotnego wpływu na konstytucję całego organizmu.
Zwiększenie ilości wolnej endotoksyny we krwi ma daleko posunięte konsekwencje patologiczne. Bardzo wysokie ogólnoustrojowe stężenia LPS, tak jak w przypadku sepsy (ostra endotoksemia) prowadzą do reakcji gorączkowych, spadku ciśnienia krwi, aktywacji krzepliwości krwi i układu uzupełniającego aż do zagrażających życiu stanów wstrząsowych. Lecz również trwałe nieznaczne podwyższenie zawartości endotoksyn we krwi, wykraczające poza zdolność detoksykacyjną wątroby skutkuje w organizmie powstaniem reakcji zapalnej, wprawdzie niskiego stopnia, lecz długotrwałej, określanej mianem Silent Inflammation, która może stanowić zwiastuna przewlekłych stanów zapalnych.
| Badania dokumentują asocjację endotoksemii z następującymi przewlekłymi schorzeniami zapalnymi: | |
|
|
LPS jest w stanie aktywować adipocyty tkanki tłuszczowej. Wydzielają one substancje posłańcze, indukujące infiltrację tkanki tłuszczowej przez prozapalne komórki odpornościowe. Równocześnie dochodzi do zablokowania przez komórki tłuszczowe czynności produkcyjnych adiponetyktyny, hormonu hamującego powstawanie stanu zapalnego. W przypadku ustawicznej stymulacji adipocytów i imigrujących makrofagów przez LPS utworzone mediatory zapalenia wywierają taki skutek, że receptory insuliny komórek tłuszczowych tracą stopniowo swą zdolność reagowania na insulinę – adipocyty stają się insulinooporne i rozwija się cukrzyca typu 2.
Oprócz wydzielania prozapalnych cytokin dalszą poważną konsekwencję pośredniczenia przez TLR4 w stymulacji komórek odpornościowych przez endotoksynę stanowi zaindukowane w ramach Silent Inflammation przez aktywowany czynnik transkrypcji NK-KB wytwarzanie reaktywnego tlenu oraz rodników azotowych. Wynikający stąd stres oksydacyjny i/lub azotowy skutkuje często dysfunkcją mitochondriów zaś z tym wiąże się zaburzenie metabolizmu energetycznego, które w rezultacie może objawiać się w postaci wyczerpania, spadku wydajności, depresji, zaburzeń koncentracji i pamięci, bólów głowy, podatności na infekcje oraz zaburzeniami krążenia.
Możliwe przyczyny endotoksemii są różnorodne. Po posiłku bogatym w tłuszcze może przejściowo dojść do wzrostu zawartości endotoksyn we krwi, gdyż LPS adsorbuje do powstających w wyniku trawienia lipidów chylomikronów i w związku z tym jest pobierany przez enterocyty i transportowany do krwi (patrz ilustracja 4: możliwość ①).
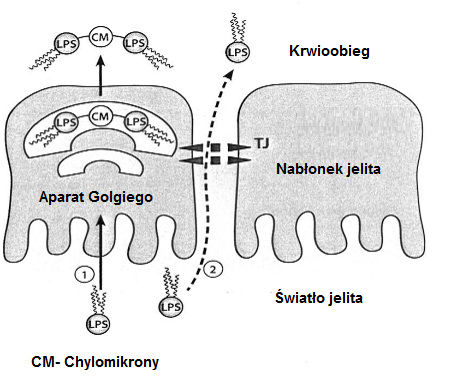
Ilustr. 4: Translokacja LPS z jelita (zmodyfikowano wg Caesar et al., 2010)
Utrzymujące się zwiększone stężenie LPS we krwi należy jednakże z reguły uznać za wskazówkę mówiącą o wystąpieniu dysfunkcji w organizmie, co wymaga dalszych badań diagnostycznych w celu wyjaśnienia patomechanizmów leżących u podłoża endoksynemii:
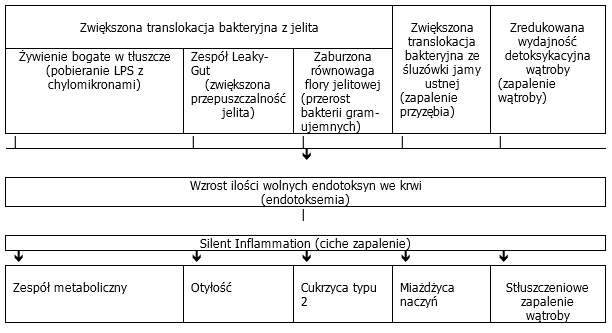
Ilustr. 5: Przyczyny i skutki endotoksemi